US Pharm. 2017;42(10):35-41.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2016 (TABLE 1) detail the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. This review is intended to be objective rather than evaluative in content. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. Studies have demonstrated the appearance of “new” adverse reactions for many NMEs within several years after they first become available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons that were not recognized at the time of approval. Therefore, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
Crisaborole (Eucrisa, Pfizer)
Indication and Clinical Profile1,2: Crisaborole is a topical agent approved for the treatment of mild-to-moderate atopic dermatitis in patients aged 2 years and older. Atopic dermatitis is a chronic inflammatory skin condition that results in pruritus and xerosis of raised red lesions that are more common on certain areas of the body depending on the patient’s age. Scratching the lesions often causes swelling, cracking, and weeping of the skin and eventually leads to skin thickening. Atopic dermatitis is the most common form of eczema, typically beginning in early childhood and lasting until adulthood. Although the exact etiology of atopic dermatitis is unknown, a combination of genetic, environmental, and immunologic factors are thought to be involved.
FDA approval of crisaborole was based on results from two randomized, multicenter, double-blind, vehicle-controlled trials in 1,522 patients aged 2 years and older with atopic dermatitis and a treatable body-surface area of 5% to 95%. The efficacy outcome was the proportion of patients achieving an Investigator’s Static Global Assessment (ISGA) score of 0 (clear) or 1 (mostly clear) at therapy day 29. A significantly greater proportion of crisaborole patients versus controls achieved an ISGA score of 0 or 1, with 32.8% versus 25.4% and 31.4% versus 18% of patients achieving the primary endpoint in studies 1 and 2, respectively.
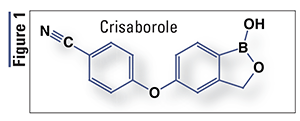
Pharmacology and Pharmacokinetics1,2: Crisaborole, a structurally unique phenoxybenzoxaborole (FIGURE 1), is an inhibitor of the enzyme phosphodiesterase (PDE) 4, specifically targeting the PDE isozyme 4B (PDE4B). PDE4B inhibition suppresses the release of tumor necrosis factor-alpha, interleukin (IL)-12, IL-23, and other cytokines believed to be involved in immune response and inflammation in disorders such as dermatitis. Also, the boron atom present in this drug enhances the lipophilicity and skin penetration necessary for activity.
Crisaborole is absorbed topically. At therapy day 8, its mean maximum plasma concentration and AUC 0 to 12 hours post dose are 127 ng/mL and 949 ng*h/mL, respectively, and it has an accumulation factor of 1.9. Crisaborole is 97% plasma protein bound and is substantially metabolized into two inactive metabolites via hydrolysis and oxidation. This agent is primarily cleared renally.
Adverse Reactions and Drug Interactions1,2: The most common adverse effect in patients receiving crisaborole in clinical trials was application-site pain. Rare but serious hypersensitivity reactions have occurred, including contact urticaria. Patients taking crisaborole should discontinue the medication if they experience severe pruritus, swelling, or erythema at the application site or a distant site, as this may be indicative of hypersensitivity. Animal-reproduction studies of crisaborole found no adverse effects at high oral doses; however, pregnant patients have not been studied, and the potential benefits and risks of crisaborole treatment should be weighed before use during pregnancy.
Crisaborole and its metabolites do not inhibit or induce any CYP450 enzyme to an extent significant enough to cause the potential for drug interactions involving CYP enzymes, and there are no other known drug interactions with crisaborole.
Dosage and Administration1,2: Crisaborole is supplied as a 2% ointment in 60-g and 100-g tubes. The drug should be applied as a thin layer twice daily to the affected areas. Crisaborole is for topical use only; it is not for ophthalmic, oral, or intravaginal use. No dose adjustments are required in patients with renal or hepatic impairment. Patients aged more than 65 years may respond differently than younger patients, but data are insufficient to compare treatment outcomes in these groups.
Ixekizumab (Taltz, Eli Lilly)
Indication and Clinical Profile3,4: Ixekizumab is approved to treat adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy and phototherapy. Psoriasis, an autoimmune disorder that occurs more commonly in persons with a family history of the disease, usually begins between the ages of 15 to 35 years. The most common form of psoriasis is plaque psoriasis, in which the patient develops thick, red skin with flaky, silver-white scales. The approval of this drug gives patients with plaque psoriasis another treatment option for the relief of the skin irritation and discomfort associated with the condition.
The safety and efficacy of ixekizumab was based on three double-blind, multicenter, phase III trials involving more than 3,800 subjects from 21 countries with moderate-to-severe plaque psoriasis. These studies, designated as UNCOVER-1, UNCOVER-2, and UNCOVER-3, compared ixekizumab (160-mg starting dose followed by 80 mg every 2 weeks) with placebo for 12 weeks. UNCOVER-2 and -3 included an additional comparator arm in which patients received etanercept (50 mg twice a week) for 12 weeks. UNCOVER-1 and -2 also evaluated response rates with ixekizumab during the maintenance period through 60 weeks. In these studies, the co-primary efficacy endpoints at 12 weeks were a 75% improvement in the composite Psoriasis Area Severity Index (PASI) score and static Physician’s Global Assessment (sPGA) score of 0 or 1 and at least a 2-point improvement from baseline. In all three studies, 87% to 90% of ixekizumab patients showed significant improvement in psoriasis plaques (PASI 75) at 12 weeks, and 81% to 83% achieved an sPGA score of 0 or 1. The majority of ixekizumab patients (68%-71%) achieved virtually clear skin (PASI 90), and 35% to 42% realized complete resolution of psoriasis plaques (PASI 100, sPGA score of 0). Among placebo patients, up to 7% achieved PASI 75 and an sPGA score of 0 or 1, and only up to 3% achieved PASI 90. Also, in UNCOVER-1 and -2, of patients who responded to ixekizumab at 12 weeks, 75% consistently maintained that response at the 60-week endpoint. Ixekizumab was statistically superior to etanercept at all skin-clearance levels, including PASI 75 and sPGA score 0 or 1 at 12 weeks. In an integrated analysis of the U.S. sites in the two active comparator studies, the respective response rates for ixekizumab versus etanercept were 87% versus 41% for PASI 75 and 73% versus 27% for sPGA score of 0 or 1. Since 2016, ixekizumab has been in phase III clinical trials for the treatment of active psoriatic arthritis.
Pharmacology and Pharmacokinetics3,4: Ixekizumab is a humanized immunoglobulin G4 (IgG4) monoclonal antibody (Mab) produced in Chinese hamster ovary cells. It consists of two light chains and two heavy chains linked by disulfide bridges. Both heavy chains are glycosylated at asparagine 296. In the hinge region, a serine is replaced by a proline to reduce formation of half-antibodies and heterodimers in the manufacturing process. The terminal lysine found in wild-type IgG4 is removed. Ixekizumab selectively binds to the interleukin (IL)-17A cytokine and inhibits its interaction with the IL-17 receptor. IL-17A is a naturally occurring cytokine that is involved in normal inflammatory and immune responses. This mechanism is similar to that of another antipsoriasis antibody, brodalumab (Siliq), which was approved in February 2017. Ixekizumab has affinity for the homodimer IL-17A and the heterodimer IL-17A/F, but not to other members of the IL-17 family.
After SC injection, ixekizumab has a bioavailability of 54% to 90%. Highest plasma concentrations are reached after 4 to 7 days following a single dose. With the usual dosing regimen (loading plus a dose every 2 weeks), steady-state concentrations are usually reached by week 8. Similar to other Mabs, ixekizumab appears to be degraded by proteolysis. Its elimination half-life is 13 days.
Adverse Reactions and Drug Interactions3,4: The most common adverse effects attributed to ixekizumab during clinical trials were upper-respiratory infections, injection-site reactions, and tinea. The overall incidence of infection in patients treated with ixekizumab was 27% compared with 23% of placebo patients. If signs or symptoms of clinically important chronic or acute infection occur or if a serious infection develops, ixekizumab should be discontinued until the infection resolves. Patients should be evaluated for tuberculosis (TB) prior to initiation of ixekizumab, and therapy should not be started in patients with active TB. Also, it is advised that all age-appropriate immunizations (per guidelines) be completed prior to initiation of ixekizumab therapy and that the use of live vaccines be avoided. Serious immune-based allergic reactions and worsening of inflammatory bowel disease were reported with the use of ixekizumab, so patients should be monitored closely for these conditions. Since ixekizumab suppresses immune function, it is marketed with a medication guide to inform patients of their greater risk of an infection or of an allergic or autoimmune condition. At present, no data are available on ixekizumab use in pregnant or breastfeeding women; however, human IgG is known to cross the placental barrier, so it is possible that the drug may be transmitted from the mother to the fetus.
No formal drug-interaction studies have been conducted with ixekizumab to date, but neither this drug nor IL-7 is known to directly interact with CYP enzymes. However, the formation of CYP enzymes can be altered by increased levels of some cytokines (e.g., IL-1, IL-6, IL-10, tumor necrosis factor-alpha, interferon) during chronic inflammation, and ixekizumab—an IL-17A antagonist—could normalize the formation of CYP enzymes. Therefore, upon initiation or discontinuation of ixekizumab in patients receiving concomitant drugs that are CYP substrates (particularly those with a narrow therapeutic index), monitoring for effect (e.g., for warfarin) or drug concentration (e.g., for cyclosporine) and dose modification of the CYP450 substrate should be considered.
Dosage and Administration3,4: Ixekizumab is supplied as an autoinjector and prefilled syringe containing 80 mg/mL of active drug for SC administration. The recommended dosage is 160 mg (two 80-mg injections) at week 0, followed by 80 mg at weeks 2, 4, 6, 8, 10, and 12, then 80 mg every 4 weeks. Ixekizumab should not be used in patients with a previous serious hypersensitivity reaction—such as anaphylaxis—to the drug product, and all patients should be evaluated for tuberculosis prior to initiation of ixekizumab treatment.
Lixisenatide (Adlyxin, Sanofi-Aventis)
Indication and Clinical Profile5,6: Lixisenatide is a once-daily injectable glucagon-like peptide-1 (GLP-1) receptor agonist that has been approved for the treatment of type 2 diabetes (DM2) in adult patients as an adjunct to diet and exercise. DM2 is a metabolic disorder characterized by insulin resistance and reduced insulin secretion that results in long-term high blood-glucose levels. Over time, high blood-glucose levels can lead to a number of complications, including retinopathy, nephropathy, neuropathy, reduced circulation, and heart disease. Lixisenatide is the fifth GLP-1 agonist to be approved by the FDA for the treatment of DM2, the others being exenatide (Byetta/Bydureon), liraglutide (Victoza), albiglutide (Tanzeum), and dulaglutide (Trulicity).
FDA approval of lixisenatide was based on data from 10 trials that evaluated lixisenatide 20 mcg once daily as standalone therapy and in combination with metformin, sulfonylureas, pioglitazone, and basal insulin in more than 5,000 subjects with DM2. The primary endpoint of all trials was a reduction in hemoglobin A1C percentage on therapy from baseline. In these trials, lixisenatide treatment resulted in significant reductions in A1C, ranging from 0.28% to 0.84%. In comparative studies versus exenatide 10 mcg twice daily, insulin glulisine once daily, and insulin glulisine thrice daily, lixisenatide was deemed noninferior to all comparators; however, treatment with exenatide thrice daily and insulin glulisine thrice daily yielded significant A1C reductions of –0.17% and –0.23%, respectively, compared to lixisenatide. A study of 6,068 patients with DM2 who were at risk for atherosclerotic cardiovascular disease found that lixisenatide did not increase the risk of cardiovascular adverse events in these patients.
Pharmacology and Pharmacokinetics5,6: Lixisenatide is a 44-amino-acid peptide with an amide group on its C terminus (FIGURE 2). It is derived from the first 39 amino acids of the peptide extendin-4, which was isolated from Gila monster venom, omitting the proline at position 38 and adding and incorporating six additional lysine residues. Lixisenatide’s therapeutic effect of lowering blood-glucose levels is elicited via agonist activity at GLP-1 receptors present in the pancreas and stomach. GLP-1 receptor activation in the pancreas stimulates glucose-dependent insulin secretion and suppresses glucagon secretion, directly lowering blood-glucose levels, as observed with other GLP-1 agonists. Furthermore, activation of GLP-1 receptors in the stomach results in delayed gastric emptying, which in turn promotes satiety and reduces appetite.

Lixisenatide achieves its peak plasma concentration 1 to 3.5 hours after injection and has a volume of distribution of 100 L. As a protein analogue, lixisenatide is presumed to undergo metabolism via proteolytic degradation and further elimination by glomerular filtration. It has a mean terminal half-life of approximately 3 hours.
Adverse Reactions and Drug Interactions5,6: The most common adverse reactions for lixisenatide reported in clinical trials were nausea, vomiting, delayed gastric emptying, headache, diarrhea, and dizziness. As with other GLP-1 agonists, use of lixisenatide has resulted in acute kidney injury, worsening of chronic renal failure, and pancreatitis. Patients should be monitored for signs and symptoms of pancreatitis and declining renal function, and the drug should be discontinued if either is suspected. Patients taking lixisenatide should be monitored for allergic reactions because severe hypersensitivity reactions including angioedema and anaphylaxis were reported in 0.1% of patients in clinical trials. During treatment, patients may develop antibodies to lixisenatide that may result in reduced efficacy and an increased incidence of allergic reactions and injection-site reactions. No adverse effects were noted in animal-reproduction studies; however, because there are no adequate studies of this drug in pregnant women, lixisenatide should be used during pregnancy only if the potential benefit outweighs the potential risk to the fetus.
Because it slows gastric emptying, concurrent use of lixisenatide with oral contraceptives may result in reduced oral-contraceptive absorption and efficacy and may delay therapeutic effects of oral antibiotics and acetaminophen. Patients using lixisenatide in combination with sulfonylureas and insulins are at increased risk for hypoglycemia; therefore, patients taking these medications may require dose reduction when lixisenatide therapy is initiated.
Dosage and Administration5,6: Lixisenatide is available as a single-patient-use, 3-mL, prefilled pen for SC injection; the starter pen contains lixisenatide 50 mcg/mL and the maintenance pen contains lixisenatide 100 mcg/mL. Recommended dosing is 10 mcg SC once daily for 14 days followed by 20 mcg SC once daily. Lixisenatide should be avoided in pediatric patients; it also should be avoided in patients with an estimated glomerular filtration rate (eGFR) <15 mL/minute/1.73 m2, as safety and efficacy in this population have not been established. No dose adjustments are required for patients with hepatic impairment or those with an eGFR 15 mL/minute/1.73 m2. However, patients with reduced renal function should be closely monitored because they are at increased risk for hypoglycemia and for gastrointestinal adverse reactions. Lixisenatide should not be used in patients with gastroparesis, as it may further slow gastric emptying.
Reslizumab (Cinqair, Teva)
Indication and Clinical Profile7,8: Asthma is a chronic disease that causes inflammation in the airways of the respiratory tract. During an asthma attack, the airways narrow, making it difficult to breathe. Severe asthma attacks often are life-threatening and result in hospitalization. According to the CDC, more than 22 million people in the United States have asthma, and more than 400,000 asthma-related hospitalizations occur each year. Reslizumab is specifically indicated for add-on maintenance treatment in patients aged 18 years and older who have a history of severe asthma attacks with an eosinophilic phenotype. Eosinophilic asthma is characterized by a higher-than-normal level of eosinophils in the respiratory tract and sputum, and it has been demonstrated that the number of eosinophils in these tissues correlates with asthma severity.
FDA approval of reslizumab was based on four double-blind, randomized, placebo-controlled trials involving more than 1,000 adult and adolescent patients with severe asthma who were on currently available therapies. Reslizumab 3 mg/kg or placebo was administered every 4 weeks as an add-on asthma treatment. Compared with placebo, reslizumab patients had reductions in asthma exacerbations of up to 59%, as well as significant improvements in lung function as measured by the volume of air exhaled in 1 second. Reslizumab patients also experienced significantly improved asthma-related quality of life.
Pharmacology and Pharmacokinetics7,8: Reslizumab is a humanized immunoglobulin G4-kappa monoclonal antibody (Mab) produced by recombinant DNA technology in murine myeloma nonsecreting 0 cells that functions as an interleukin-5 (IL-5) antagonist. IL-5 is the major cytokine responsible for the differentiation, recruitment, activation, and survival of eosinophils, mediators that contribute to inflammation in respiratory tissue. Reslizumab binds to IL-5 with high affinity (Kd 81 pM) and inhibits IL-5 signaling, which reduces the production and survival of eosinophils. In all trials, reductions in blood eosinophil counts were observed following the first dose of reslizumab and were maintained through the treatment period and until reslizumab was discontinued.
Peak serum concentrations of reslizumab occur at the end of infusion and decline in a biphasic manner. The mean observed accumulation ratio of reslizumab following multiple doses of administration ranges from 1.5-fold to 1.9-fold. Reslizumab has a volume of distribution of approximately 5 L, suggesting minimal distribution to the extravascular tissues. Its clearance is approximately 7 mL/hour, and it has a half-life of about 24 days. Similar to other Mabs, reslizumab is degraded by enzymatic proteolysis into small peptides and amino acids. As reslizumab binds to a soluble target, it does not appear to go through a target-mediated clearance. Systemic exposure to reslizumab seems to be unaffected by the presence of treatment-emergent antireslizumab antibodies. Population studies have demonstrated no significant variations in reslizumab pharmacokinetics based on age, race, or sex. These studies also suggest no significant difference in the pharmacokinetics of reslizumab in patients with normal liver- or kidney-function tests versus those with mild hepatic impairment or mild-to-severe renal impairment.
Adverse Reactions and Drug Interactions7,8: The most common adverse effect of reslizumab in clinical trials was oropharyngeal pain. In clinical trials, oropharyngeal pain occurred in 2% of subjects, and elevated baseline creatine phosphokinase was more common in subjects receiving reslizumab versus placebo. Myalgia and some musculoskeletal adverse reactions also were reported more often in reslizumab-treated subjects than in subjects taking placebo. Reslizumab can cause serious side effects, including hypersensitivity reactions (0.3% of the trial population), which may be life-threatening and require drug discontinuation. Hypersensitivity symptoms, which include dyspnea, decreased oxygen saturation, wheezing, vomiting, and skin and mucosal involvement (including urticarial), are typically observed as early as 20 minutes after completion of infusion; reslizumab therefore has a black box warning regarding the potential for anaphylaxis. Malignancies, which were diverse in nature and did not involve clustering of any particular tissue type, were observed in several subjects in clinical trials (0.6%). Also, because eosinophils are involved in the normal immunologic response to some helminth infections, patients with preexisting helminth infections should be cleared of infection before reslizumab therapy is initiated. If a patient becomes infected while receiving reslizumab and does not respond to anti-helminth treatment, reslizumab should be discontinued until the parasitic infection resolves.
No formal clinical drug-interaction studies have been conducted with reslizumab. In vitro data indicate that IL-5 and reslizumab are unlikely to affect CYP1A2, -2B6, or -3A4 enzyme activity. Population studies indicate that concomitant use of other drugs, including leukotriene antagonists and corticosteroids, does not affect the pharmacokinetics of reslizumab. It should be noted that systemic or inhaled corticosteroid therapy should not be discontinued upon initiation of reslizumab therapy. A reduction in corticosteroid dose may be associated with systemic withdrawal symptoms or unmask conditions previously suppressed by systemic corticosteroid therapy. If appropriate, reductions in corticosteroid dose should be gradual and performed under the supervision of a physician.
Dosage and Administration7,8: Reslizumab is supplied as a single-use, preservative-free solution for IV administration with 100 mg of drug in a 10-mL glass vial (10 mg/mL). The recommended dosage is 3 mg/kg once every 4 weeks administered over 20 to 50 minutes. The drug should be administered by a healthcare professional in a clinical setting capable of managing anaphylaxis.
REFERENCES
1. Eucrisa (crisaborole) package insert. Palo Alto, CA: Anacor Pharmaceuticals, Inc; December 2016.
2. Nazarian R, Weinberg JM. AN-2728, a PDE4 inhibitor for the potential topical treatment of psoriasis and atopic dermatitis. Curr Opin Investig Drugs. 2009;10:1236-1242.
3. Taltz (ixekizumab) package insert. Indianapolis, IN: Eli Lilly and Co; March 2016.
4. Mease PJ, van der Heijde D, Ritchlin CT, et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis. 2017;76:79-87.
5. Adlyxin (lixisenatide) package insert. Bridgewater, NJ: Sanofi-Aventis U.S. LLC; July 2016.
6. Elkinson S, Keating GM. Lixisenatide: first global approval. Drugs. 2013;73:383-391.
7. Cinqair (reslizumab) package insert. Frazer, PA: Teva Pharmaceutical Industries, Ltd; March 2016.
8. Castro M, Zangrilli J, Wechsler ME, et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir Med. 2015;3:355-366.
To comment on this article, contact rdavidson@uspharmacist.com.



