US Pharm. 2018;(43):HS-7-HS-12.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2017-2018 (TABLE 1) detail the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. For example, “new” adverse reactions for many NMEs often emerge within 2 to 3 years after they first become available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons that were not recognized at the time of approval. Therefore, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
Angiotensin II (Giapreza, La Jolla)
Indication and Clinical Profile1,2: Giapreza is a synthetic formulation of angiotensin II that has been approved with a specific indication to increase blood pressure in adults with septic or other distributive shock. Distributive shock is a serious medical condition characterized by excessive vasodilation leading to an inability to maintain adequate blood flow to vital tissues. The reduced blood flow results in inadequate perfusion to these tissues, which ultimately can lead to organ dysfunction, organ failure, and death. Giapreza was FDA approved under priority review and is the first synthetic angiotensin II to be approved by the FDA.
FDA approval of synthetic angiotensin II was based on data from a double-blind, placebo-controlled study in 321 subjects with septic or other distributive shock who remained hypotensive despite fluid and vasopressor therapy. The primary endpoint was the percentage of subjects who achieved either a mean arterial pressure (MAP) of ≥75 mmHg or a ≥10-mmHg increase in MAP without an increase in baseline vasopressor therapy at 3 hours. A statistically higher percentage of patients treated with synthetic angiotensin II achieved the primary endpoint compared with those receiving placebo (70% vs. 23%). There was no significant difference in mortality through day 28 posttreatment between the active and control groups (46% and 54%, respectively).
Pharmacology and Pharmacokinetics1,2: Synthetic angiotensin II (Figure 1) binds to G protein–coupled angiotensin II type 1 receptors on vascular smooth- muscle cells, causing smooth-muscle contraction (vasoconstriction) and an overall increase in blood pressure. Synthetic angiotensin II achieves its therapeutic effect at a median of 5 minutes postinfusion and has a half-life of less than 1 minute. It is metabolized throughout the body by ACE 2 to angiotensin III or by aminopeptidase A to angiotensin-(1-7), metabolites that produce reduced and opposite effects, respectively, of angiotensin II.

Adverse Reactions and Drug Interactions1,2: The most common adverse reactions (≥5%) reported by patients receiving synthetic angiotensin II in clinical trials were thromboembolic events, thrombocytopenia, tachycardia, fungal infection, delirium, and acidosis. Patients receiving synthetic angiotensin II are at increased risk for arterial and venous thrombotic and thromboembolic events. To reduce this risk, patients receiving synthetic angiotensin II should receive concurrent venous thromboembolism prophylaxis. Animal-reproduction studies have not been conducted with synthetic angiotensin II, and there are insufficient published data to determine an association between angiotensin II use in pregnant women and the risk of adverse fetal outcomes. Because of their impact on angiotensin II metabolism, the concomitant use of synthetic angiotensin II with ACE inhibitors may result in an increase in synthetic angiotensin II therapeutic effects. These therapeutic effects may be decreased when the drug is used in combination with angiotensin II receptor blockers.
Dosage and Administration1,2: Synthetic angiotensin II is supplied as a solution for IV infusion in single-dose, 1-mL, and 2-mL vials at a concentration of 2.5 mg/mL. Prior to administration, one vial of synthetic angiotensin II must be diluted in 0.9% saline to achieve a final concentration of 5,000 ng/mL, or 10,000 ng/mL for fluid-restricted patients. The recommended starting dosage is 20 ng/kg/min via continuous IV infusion through a central venous line. Monitor blood pressure and titrate synthetic angiotensin II every 5 minutes in increments of up to 15 ng/kg/min as needed to achieve or maintain target blood pressure. The maximum recommended dosage is 80 ng/kg/min during the first 3 hours of treatment, and 40 ng/kg/min during maintenance dosing. Based on blood pressure, synthetic angiotensin II should be down-titrated every 5 to 15 minutes using up to 15 ng/kg/min increments once the underlying shock has resolved. No dosing adjustments are required in renal or hepatic impairment. Giapreza has not been studied in pediatric patients.
Ibalizumab-uiyk (Trogarzo, Theratechnologies)
Indication and Clinical Profile3,4: Ibalizumab-uiyk is a CD4-directed postattachment HIV-1 inhibitor that, when used with other antiretroviral therapy (ART), has been approved for the treatment of HIV type 1 (HIV-1) infection in heavily treatment-experienced adults with multidrug-resistant HIV-1 infection who are failing their current ART regimen. HIV is a retrovirus that affects various immune cells and, over time, can lead to progressive immunodeficiency and life-threatening opportunistic infections. It is estimated that up to 25,000 Americans have multidrug-resistant HIV; roughly half of them have failed their current ART regimen, resulting in transmittable multidrug-resistant HIV. Owing to the need for new therapies for these patients, ibalizumab-uiyk received priority-review status from the FDA and was designated as a breakthrough therapy and an orphan drug.
FDA approval of ibalizumab-uiyk was based on data from a single-arm, multicenter clinical trial of 40 HIV-1–infected subjects who had documented multidrug-resistant HIV-1; had a viral load (VL) >1,000 copies/mL and documented resistance to at least one nucleoside reverse transcriptase inhibitor (NRTI), one nonnucleoside reverse transcriptase inhibitor (NNRTI), and one protease inhibitor (PI); and were failing or had recently failed therapy. The primary efficacy endpoint was the proportion of subjects achieving a ≥0.5 log10 decrease in VL after 7 days of ibalizumab-uiyk therapy (functional monotherapy period), compared with those achieving the same VL 7 days prior to starting ibalizumab-uiyk (control period). Upon completion of the functional monotherapy period, 83% of patients achieved the target VL reduction, compared with 3% of patients who achieved the same goal during the control period. After the functional monotherapy period, ibalizumab-uiyk was continued in combination with optimized background ART in all patients for a total duration of 25 weeks, at which point 55% and 48% of subjects had a ≥1 log10 and ≥2 log10 reduction in VL, respectively, and 50% of patients achieved a VL of <200 HIV-1 RNA copies/mL.
Pharmacology and Pharmacokinetics3,4: Ibalizumab-uiyk is a recombinant humanized monoclonal antibody that binds to domain 2 of CD4, functionally blocking HIV-1 from infecting CD4+ T cells by interfering with postattachment steps required for HIV-1 entry into the host cell. It shows no antagonism toward other classes of ART, including CCR5 coreceptor antagonists, NNRTIs, NRTIs, and PIs.
Ibalizumab-uiyk exhibits nonlinear pharmacokinetics and has a volume of distribution of 4.8 L. When the infused dose is increased from 0.3 to 25 mg/kg, its clearance decreases from 9.54 to 0.36 mL/h/kg and its elimination half-life increases from 2.7 to 64 hours. When the recommended dosing regimen is followed, ibalizumab-uiyk reaches steady-state levels after the first maintenance dose with mean concentrations over 30 mcg/mL.
Adverse Reactions and Drug Interactions3,4: The most common adverse reactions (≥5%) reported by patients receiving ibalizumab-uiyk in clinical trials were diarrhea, dizziness, nausea, and rash. Patients also experienced a variety of lab abnormalities, the most common of which (≥5%) included elevations of serum bilirubin, creatinine, lipase, leukocytes, and neutrophils. The occurrence of immune reconstitution inflammatory syndrome was reported in one patient receiving ibalizumab-uiyk during clinical trials, and patients should be monitored for signs and symptoms of inflammatory response during therapy initiation. As a monoclonal antibody, ibalizumab-uiyk has the potential to be transmitted across the placenta; however, adequate data on ibalizumab-uiyk use in pregnancy are not available and animal-reproduction toxicology studies have not been conducted. Healthcare providers with pregnant patients receiving ibalizumab-uiyk are encouraged to register patients in the pregnancy-exposure registry, which monitors pregnancy outcomes. Ibalizumab-uiyk has no expected drug-drug interactions based on its mechanism of action and target-mediated drug disposition.
Apalutamide (Erleada, Janssen)
Indication and Clinical Profile5,6: Apalutamide is the first FDA-approved drug for the treatment of patients with nonmetastatic, castration-resistant prostate cancer. This is a form of prostate cancer that continues to grow despite treatment with hormone therapy. According to the National Cancer Institute (NCI), prostate cancer is the second most common type of cancer in men in the United States. The NCI estimates that over 160,000 men were diagnosed with prostate cancer in 2017, and 26,730 were expected to die of the disease. Approximately 10% to 20% of prostate cancer cases are castration-resistant, and up to 16% of these patients show no evidence that the cancer has spread at the time of the castration-resistant diagnosis. The application for this drug was granted priority review by the FDA; the agency’s goal is to take action on an application within 6 months when it determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing, or preventing a serious condition.
The safety and efficacy of apalutamide was based on the SPARTAN study, a phase III, randomized trial of 1,207 patients with nonmetastatic, castration-resistant prostate cancer. Trial subjects were randomized 2:1 to receive either apalutamide orally at a dosage of 240 mg once daily (n = 806) or placebo once daily (n = 401). All subjects received a concomitant gonadotropin-releasing hormone (GnRH) analogue or had a bilateral orchiectomy. Apalutamide decreased the risk of distant metastasis or death by 72% compared with placebo. The median metastasis-free survival was 40.5 months for apalutamide compared with 16.2 months for placebo, a difference of about 2 years. The major efficacy outcome was supported by statistically significant improvements in the following secondary endpoints: time to metastasis (TTM), progression-free survival (PFS), and time to symptomatic progression. The median TTM was 40.5 months for apalutamide compared with 16.6 months for placebo, and the median PFS was 40.5 months compared with 14.72 months for placebo.
Pharmacology and Pharmacokinetics5,6: Apalutamide (Figure 2) is an androgen receptor (AR) inhibitor that binds directly to the ligand-binding domain of the AR. By this mechanism, the drug inhibits AR nuclear translocation, inhibits DNA binding, and impedes AR-mediated transcription, resulting in inhibition of tumor growth.

Apalutamide is well absorbed (100% bioavailability), with a time to achieving peak plasma concentration (Tmax) of about 2 hours. Administration of the drug under fasting conditions or with a high-fat meal does not alter absorption. The apparent volume of distribution of apalutamide at steady state is approximately 276 L, and both the parent drug and its primary metabolite, N-desmethyl, are approximately 95% bound to plasma proteins. Metabolism by CYP2C8 and CYP3A4 is the main route of elimination. Metabolism yields an active metabolite, N-desmethyl apalutamide. Apalutamide represents 45% and N-desmethyl apalutamide represents 44% of the total AUC. Both the drug and its metabolite are eliminated by renal (65%) and biliary (24%) mechanisms. The clearance over bioavailability of apalutamide was 1.3 L/h after single dosing and increased to 2.0 L/h at steady state after once-daily dosing, likely due to CYP3A4 autoinduction. The mean effective half-life of apalutamide is approximately 3 days at steady state.
Adverse Reactions and Drug Interactions5,6: Commonly reported adverse effects of apalutamide in clinical trials included fatigue, hypertension, rash, diarrhea, nausea, weight loss, arthralgia, hot flush, decreased appetite, and peripheral edema. More severe but less common side effects of apalutamide include falls, fractures, and seizures.
Coadministration of strong CYP2C8 or CYP3A4 inhibitors is predicted to increase the exposure of N-desmethyl-apalutamide. Although no initial dose adjustment is necessary, dose reductions of apalutamide may be required for tolerability. Mild or moderate inhibitors of CYP2C8 or CYP3A4 are not expected to affect exposure. Apalutamide is a strong inducer of CYP3A4 and CYP2C19 and a weak inducer of CYP2C9 in humans. Therefore concomitant use of apalutamide with medications that are primarily metabolized by these CYP isozymes can result in lower exposure to these medications. Substitution for these medications is recommended when possible; evaluate for loss of activity if medication is continued. Concomitant administration of apalutamide with medications that are substrates of uridine diphosphate-glucuronosyl transferase (UGT) can result in decreased exposure and potential loss of efficacy. Therefore, caution should be exercised if UGT substrates must be coadministered with apalutamide. Apalutamide is a weak inducer of P-glycoprotein, breast cancer resistance protein, and organic anion-transporting polypeptide 1B1. Therefore, caution should be exercised with the concomitant use of apalutamide with medications that are substrates of these transporters because apalutamide induction can result in lower exposure of these medications and loss of efficacy.
Dosage and Administration5-6: Apalutamide is supplied as 60-mg film-coated tablets for oral administration. The recommended dosage of apalutamide is 240 mg (four 60-mg tablets) administered orally once daily, and tablets should be swallowed whole. Apalutamide may be taken with or without food. Patients should also receive a GnRH analogue concurrently or should have had a bilateral orchiectomy. If a patient experiences Grade ≥3 toxicity or an intolerable side effect, dosing should be held until symptoms improve to Grade ≤1 or original grade, then resume at the same dosage or a reduced dosage (180 mg or 120 mg), if warranted.
Acalabrutinib (Calquence, AstraZeneca)
Indication and Clinical Profile7,8: Acalabrutinib was granted accelerated approval for the treatment of adult patients with mantle cell lymphoma who have received at least one prior therapy. Mantle cell lymphoma is a rare and fast-growing type of non-Hodgkin lymphoma and, according to the National Cancer Institute, represents 3% to 10% of all non-Hodgkin lymphoma cases in the United States. It is a cancer of the lymph system, which is part of the body’s immune system and is made up of lymph tissue, lymph nodes, the spleen, thymus, tonsils, and bone marrow. By the time mantle cell lymphoma is diagnosed, it usually has spread to the lymph nodes, bone marrow, and other organs. The FDA granted this application accelerated-approval, priority- review, and breakthrough-therapy designations, along with orphan-drug designation.
The accelerated approval of acalabrutinib was based on data from a single-arm trial that included 124 patients with mantle cell lymphoma who had received at least one prior treatment. Acalabrutinib was administered orally at 100 mg twice daily until disease progression or unacceptable toxicity. Tumor response was assessed according to the Lugano classification for non-Hodgkin lymphoma. The major efficacy outcome of the trial was overall response rate (ORR) and the median follow-up was 15.2 months. The ORR, which measured how many patients experienced complete or partial shrinkage of their tumors, was 81%, and 40% of patients showed a complete response. Acalabrutinib was granted accelerated approval based on ORR. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
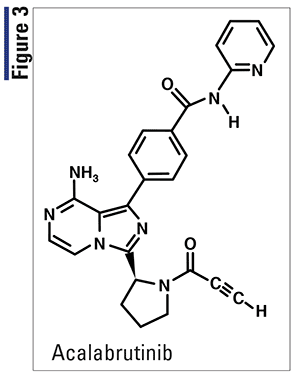
Pharmacology and Pharmacokinetics7,8: Acalabrutinib (Figure 3) is a small-molecule Bruton’s tyrosine kinase (BTK) inhibitor. Acalabrutinib and its active metabolite, ACP-5862, form a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK enzymatic activity. BTK is a signaling molecule of the B-cell antigen receptor and cytokine- receptor pathways. In B cells, BTK signaling results in activation of pathways necessary for B-cell proliferation, trafficking, chemotaxis, and adhesion.
The absolute bioavailability of acalabrutinib is 25%, with a median time to peak plasma concentrations (Tmax) of 0.75 hours. Food does not appear to significantly alter drug availability. Acalabrutinib is 97.5% bound to plasma protein and the volume of distribution is approximately 34 L. This drug is metabolized primarily by CYP3A enzymes and, to a lesser extent, by glutathione conjugation and amide hydrolysis. ACP-5862 was identified as the major active metabolite in plasma, with a geometric mean exposure (AUC) that was approximately two-to three-fold higher than the exposure of acalabrutinib. ACP-5862 is approximately 50% less potent than acalabrutinib with regard to BTK inhibition. The majority of the oral dose (84%) is excreted as metabolites in the feces; only 12% of the dose is recovered in the urine. The median terminal elimination half-life (t1/2) of acalabrutinib is less than 1 hour, whereas the active metabolite has a half-life of almost 7 hours.
Adverse Reactions and Drug Interactions7,8: In trials to date, the most common adverse effects associated with acalabrutinib were headache, diarrhea, bruising, fatigue, myalgia, anemia, thrombocytopenia, and neutropenia. More serious side effects included hemorrhage, infections, and atrial fibrillation. Grade 3 or higher bleeding events, including gastrointestinal, intracranial, and epistaxis, have been reported in 2% of patients. Overall, bleeding events, including bruising and petechiae of any grade, occurred in approximately 50% of patients with hematologic malignancies. Acalabrutinib may further increase the risk of hemorrhage in patients receiving antiplatelet or anticoagulant therapies, and patients should be monitored for signs of bleeding. Consider the benefit versus risk of withholding acalabrutinib for 3 to 7 days pre- and postsurgery, depending upon the type of surgery and the risk of bleeding. Grade 3 or higher infections occurred in 18% of trial patients. The most frequently reported Grade 3 or 4 infection was pneumonia. Infections due to hepatitis B virus reactivation and progressive multifocal leukoencephalopathy were also reported. Patients should be monitored for signs and symptoms of infection and treat as medically appropriate. Additional cancers, known as second primary malignancies, occurred in some patients taking acalabrutinib. The most frequent second primary malignancy was skin cancer, reported in 7% of patients; therefore, patients taking this drug should take measures to protect themselves from sun exposure. Women who are breastfeeding should not take acalabrutinib because it may cause harm to the newborn baby.
Avoid coadministration of acalabrutinib with strong CYP3A4 inhibitors. If a strong CYP3A4 inhibitor will be used short-term (such as anti-infectives for up to 7 days), interrupt acalabrutinib. When acalabrutinib is coadministered with a moderate CYP3A inhibitor, reduce the acalabrutinib dosage to 100 mg once daily. Also, to avoid therapeutic failure, concurrent administration of acalabrutinib with a strong CYP3A4 inducer should be avoided. If concurrent use with a CYP3A4 inducer is necessary, acalabrutinib dosages should be increased to 200 mg twice daily. If treatment with a gastric–acid reducing agent is required in patients on acalabrutinib therapy, an H2-receptor antagonist or an antacid should be considered and dosing separated by at least 2 hours. Coadministration of acalabrutinib with a proton pump inhibitor should be avoided owing to the long-lasting effect of proton pump inhibitors.
Dosage and Administration7,8: Acalabrutinib is supplied as 100-mg capsules for oral administration. The capsule should be swallowed whole with water and with or without food. The recommended dosage is 100 mg orally approximately every 12 hours until disease progression or unacceptable toxicity. Recommended dose modifications of acalabrutinib for Grade 3 or greater adverse reactions are provided in the manufacturer’s literature.
REFERENCES
1. Giapreza (angiotensin II) package insert. San Diego, CA: La Jolla Pharmaceutical Co; December 2017.
2. Kaufman MB. Pharmaceutical approval update March 2018. P & T: 2018;43(3):141-170.
3. Trogarzo (ibalizumab-uiyk) package insert. Montréal, Quebec: Theratechnologies Inc; May 2018.
4. Jacobson JM, Kuritzkes DR, Godofsky E, et al. Safety, pharmacokinetics, and antiretroviral activity of multiple doses of ibalizumab (formerly TNX-355), an anti-CD4 monoclonal antibody, in human immunodeficiency virus type 1-infected adults. Antimicrob Agents Chemother. 2009;53(2):450-457.
5. Erleada (apalutamide) package insert. Horsham, PA: Janssen Products, LP; February 2018.
6. Bossi A, Dearnaley, D, McKenzie, M, et al. ATLAS: a phase 3 trial evaluating the efficacy of apalutamide (ARN-509) in patients with high-risk localized or locally advanced prostate cancer receiving primary radiation therapy. Ann Oncol. 2016;7(suppl 6).
7. Calquence (acalabrutinib) package insert. Wilmington, DE: AstraZeneca Pharmaceuticals LP; October 2017.
8. Wu J, Zhang M, Liu, D. Acalabrutinib (ACP-196): a selective second-generation BTK inhibitor. J Hematol Oncol. 2016:9;9:21.
To comment on this article, contact rdavidson@uspharmacist.com.



